This is the main help page on how to search RNA Editing sites in REDIdb and use the embedded tools.
Searching RNA Editing sites
Browsing RNA Editing sites
REDIdb scripts download
Searching into REDIdb is very intuitive and also users with no bioinformatics skills can perform accurate searches into our database. RNA editing sites can be retrieved by organism, by location or by gene.
All these fields can be combined to get selective queries. Final result can be further filtered for "RefSeq", "Exons", "Full Orfs" by selecting their respective checkboxes on the query form.
Note: If the user does not make any choice, the search will be performed against the ENTIRE DATABASE.
RNA editing events by organism can be retrieved by selecting one of the voices in the Organism field.
Search can be done also for organelle by choosing mitochondrion/chloroplast in the Location field.
If the user is interested in all the editing events occurring in a specific gene, also searching by genename is possible.
Once the user makes his choice, the search can be further refined using selected filters.
Note: Filters can be variously combined according to the user's preferences.
The following options are admitted:
| Filter | Name | Effects |
|---|---|---|
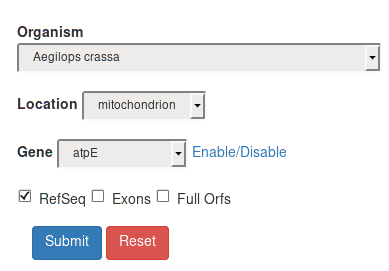 |
RefSeq |
If the RefSeq square is checked, the final results will include ONLY records from the non-redundant NCBI Reference Sequence Database. |
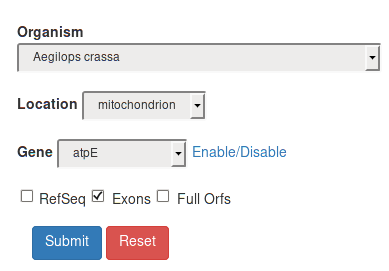 |
Exons |
By selecting the Exons filter, the final result will also include exons. Note: Exons are available if present in the Genbank flatfile from which the Redidb record was obtained. Editing events are numbered according to the exonic order. |
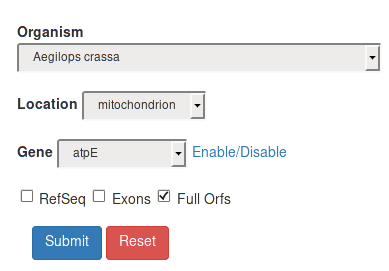 |
Full Orfs |
Trough the Full Orfs filter the query will take into account only complete genes. |
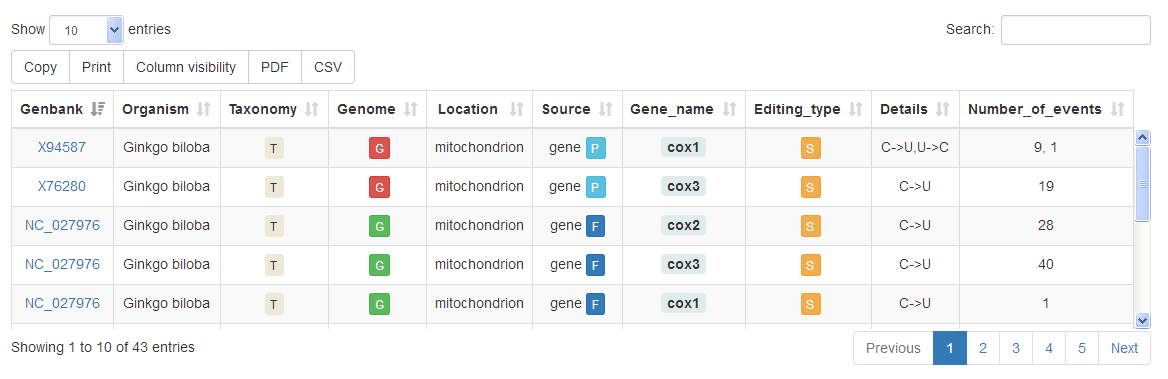
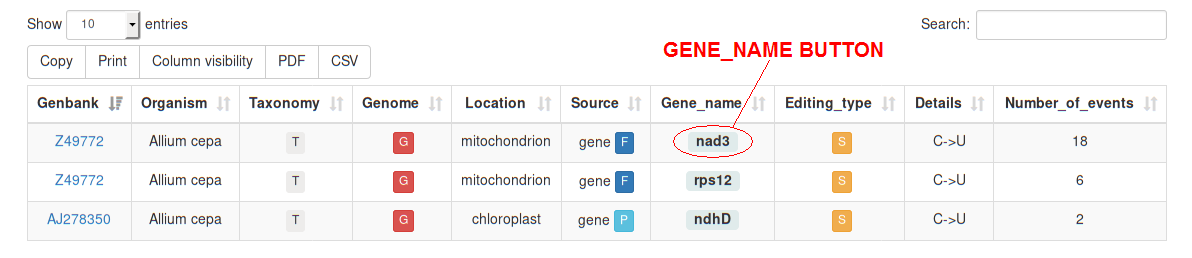
Once a query has been submitted, the corresponding results will be displayed in a Table Report including the following columns:
| Column Name | Description |
|---|---|
| Genbank | The Genbank ACCESSION number of the flatfile from which the editing event/s has/have been extracted. By clicking on this number the user will be redirected to the Genbank record's page. |
| Organism | The NAME (genus and species) of the organism corresponding to that accession number. The binomial nomenclature is the same adopted by the NCBI Taxonomy database. |
| Taxonomy | The relevant TAXONOMY according to the NCBI Taxonomy database.
By clicking on the button a circular phylogenetic tree based on jsPhyloSVG is displayed. (See the following section for further details) |
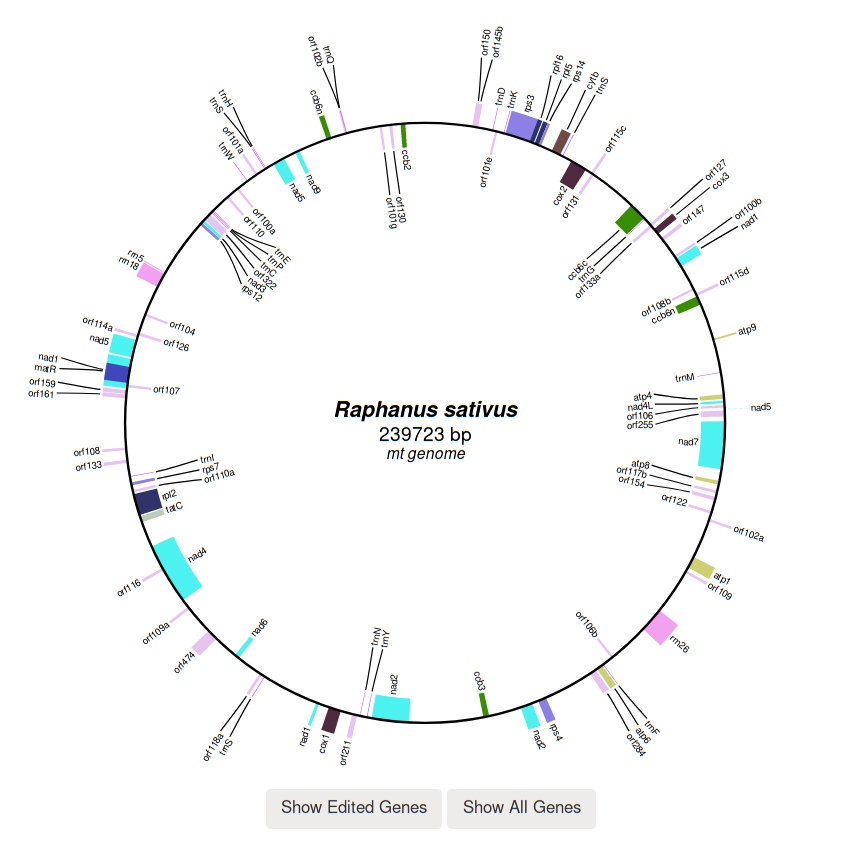
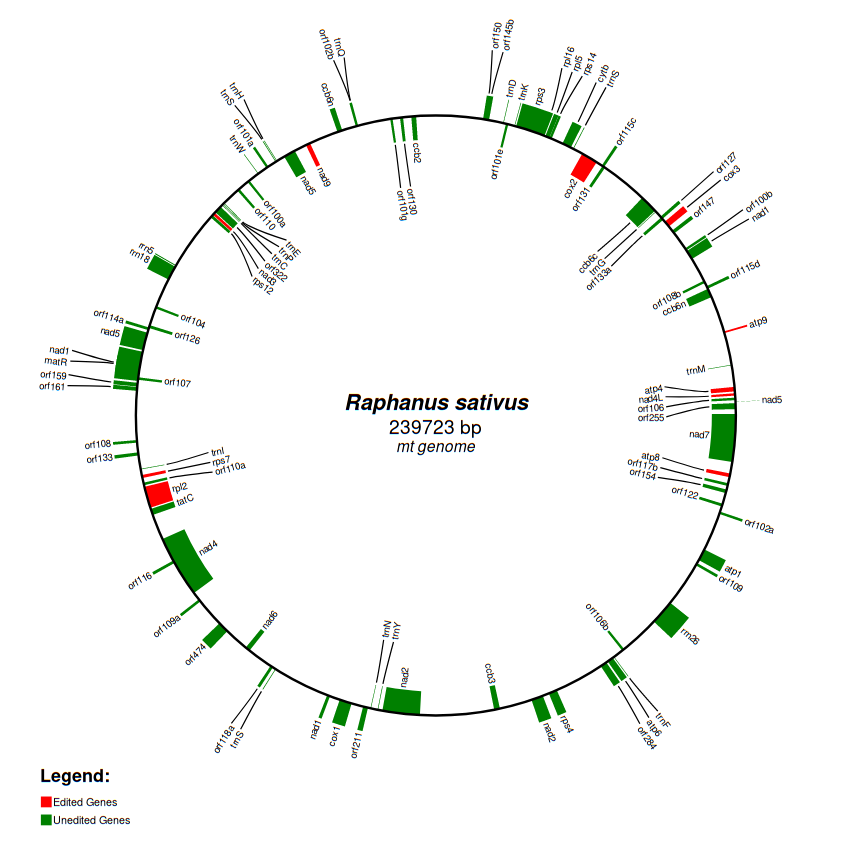
| Genome | If the COMPLETE GENOMIC SEQUENCE is available on GenBank, a is shown, otherwise a . By clicking on the button the complete genome is graphically rendered, allowing the user to visualize the reciprocal order of the edited genes. (See below for more details.) |
| Location | The CELLULAR LOCALIZATION of the editing event/s (mitochondrion or chloroplast) |
| Source | The MOLECULAR SOURCE of the record (gene, tRNA, ecc.). If the annotated sequence is complete the molecular source is followed by an , otherwise by a . |
| Gene name | The GENE NAME.
Please consider that some genes might be called in different ways and thus, before submitting a query on REDIdb,
check for gene aliases according to this page.
By clicking on the gene_name button the REDIdb RECORD PAGE relative to each entry will be shown. (See below for more details) |
| Editing type | THE TYPE OF THE OCCURRING EDITING PROCESS. It can assume the following values: "Substitution" , "Insertion" or "Deletion" , according to the fact that a specific sequence can be subjected to different RNA editing events at the same time. |
| Details | DETAILS contains all specific details for each editing type. In general, in the case of substitutions, they are showed as "genomic nucleotide-->modified cDNA nucleotide". For example, C to U or U to C substitutions are represented as "C-->U" or "U-->C", respectively. |
| Number of events | NUMBER OF EVENTS contains the total number of editing events for each editing type. The numbers are separated by commas in the case of different editing types. |
All the data contained in the Table Report can be copied/pasted (1),
printed (2), rearranged by hiding certain columns (3), or exported as
Portable Document File) (4) or Comma Separated Values)
(5). The latter format can be easily opened with spreadsheet softwares like
LibreOffice® or Ms Excel®.
The number of rows displayed for each table can be set starting from 10 up to 100 entries (6);
moreover, a search field (7) on the right corner of the page, allows the user to reduce
the rows only to them matching a specific term.

By clicking on entries gene_name button the REDIdb RECORD PAGE will appear.


Note. By clicking on the GO Accession Number (e.g. GO:0045156) the user will be redirected to the AMIGO® page relative to that GO term.
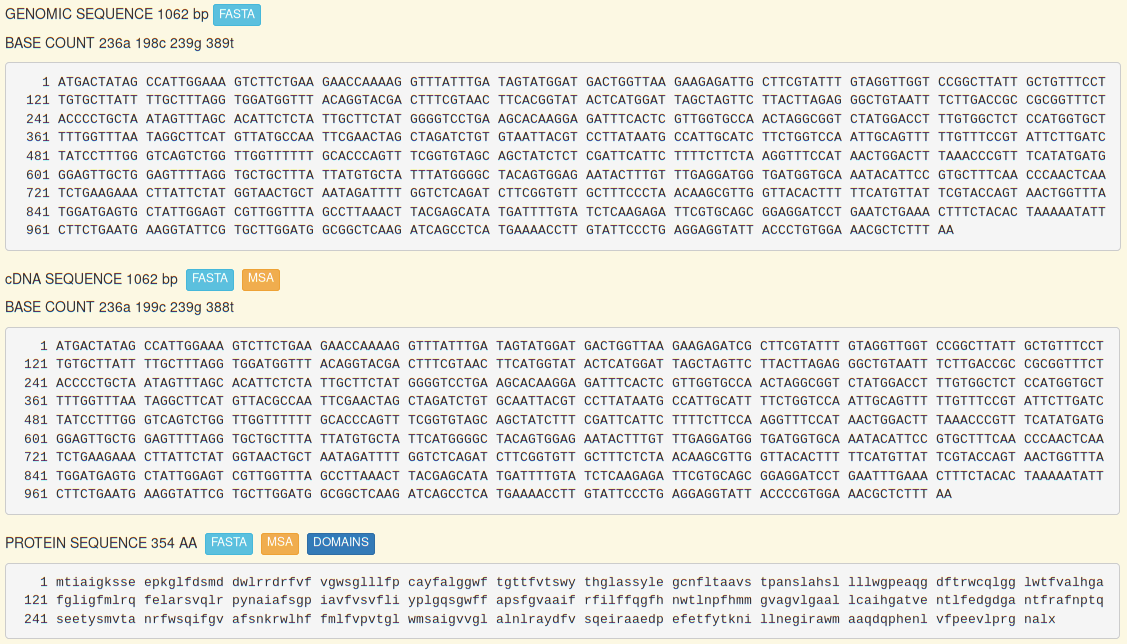
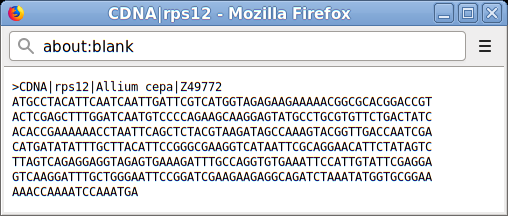
Each sequence (genomic, edited transcript, protein) can be downloaded in a Fasta-like format by clicking on the button.
A major novelty in REDIdb is the inclusion of multiple-alignments between
orthologous sequences. This aspect is very useful for assessing the editing conservation across the species annotated
in the database.
Multiple sequence alignment for each gene (or protein) is generated by using the iterative
progressive algorithm embedded with ClustalOmega®.
The graphic representation of the multi-alignment is achieved by means of
MSAViewer® a fast and lightweight Biojavascript component.
By clicking on the button near the transcript or the protein sequence,
the multiple alignment relative to orthologous of that sequence will be shown in a dedicated page.
Note:The button is available for complete sequences stored
in the database.
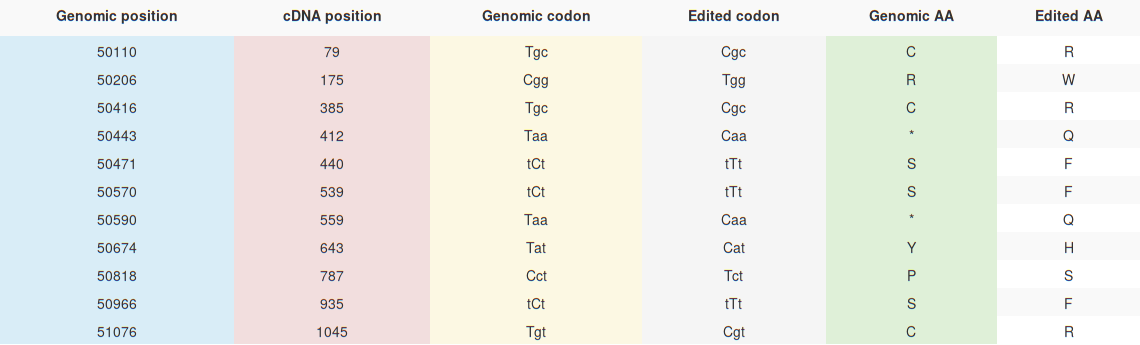
Each MSA box (shown below) contains:
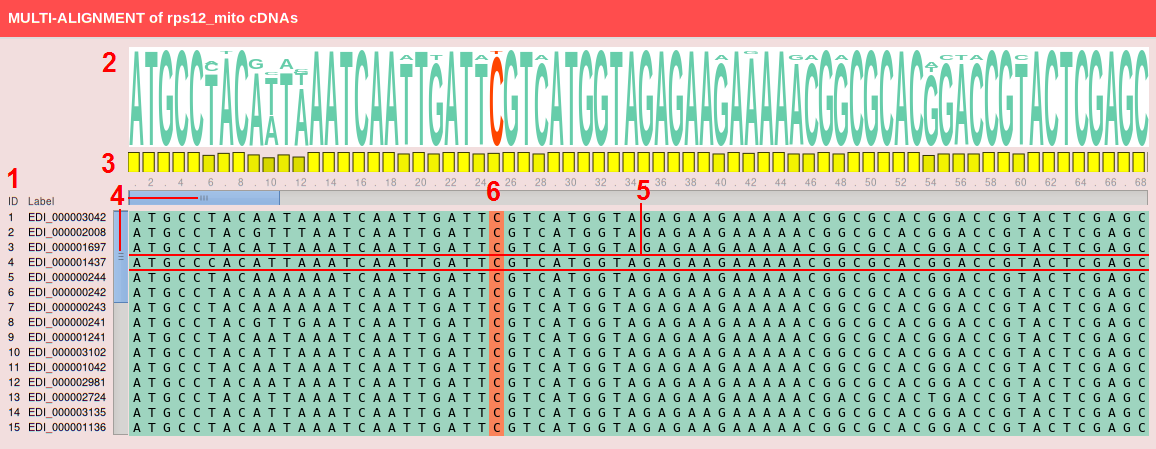

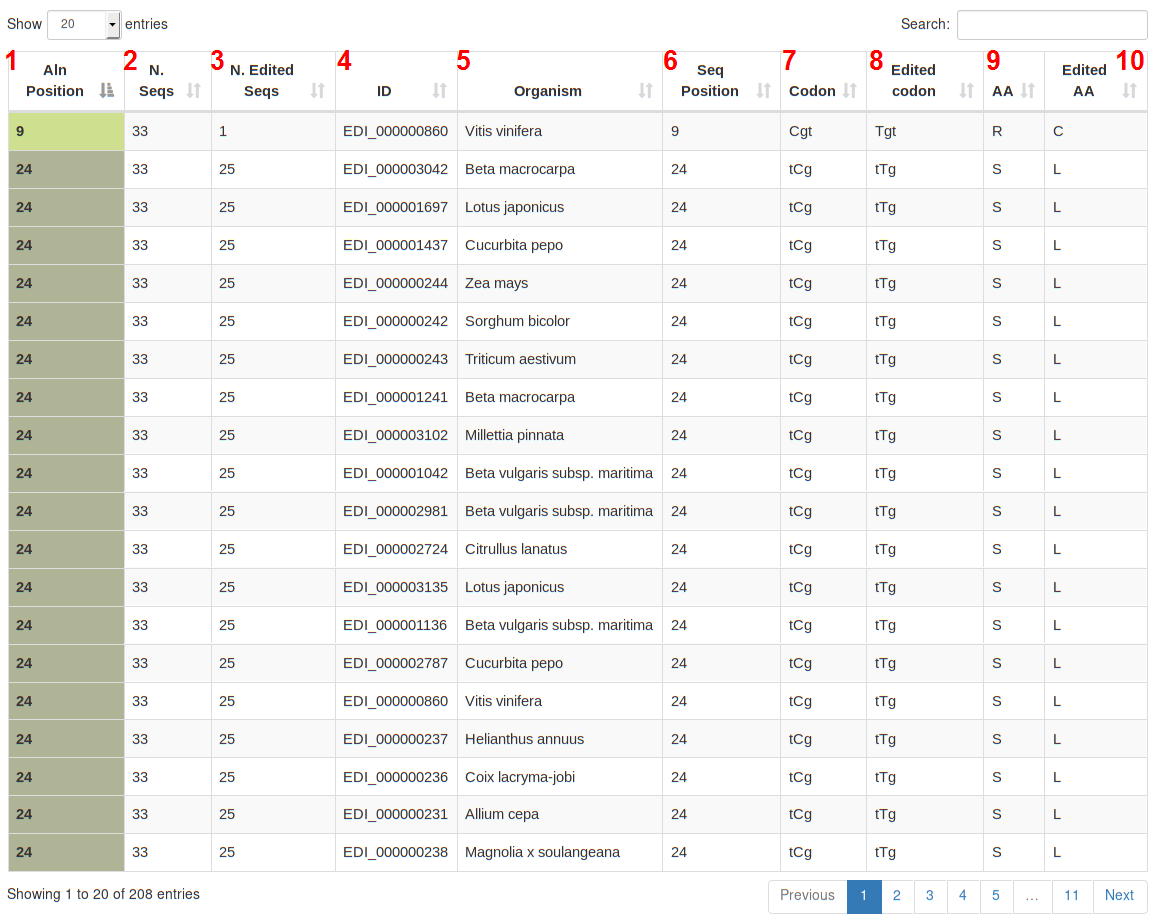
Under each MSA box a table describes more in details each RNA editing site considering its position in the multialignment. The table contains the following columns:
Sequences sharing the same editing position are coloured in the same manner on the first column. (e.g Aln 24).
Like the other REDIdb tables, the user can select how many rows wants to show per page.
A search filter on the right corner of the table, can also be used to reduce the rows
only to them matching specific criteria.
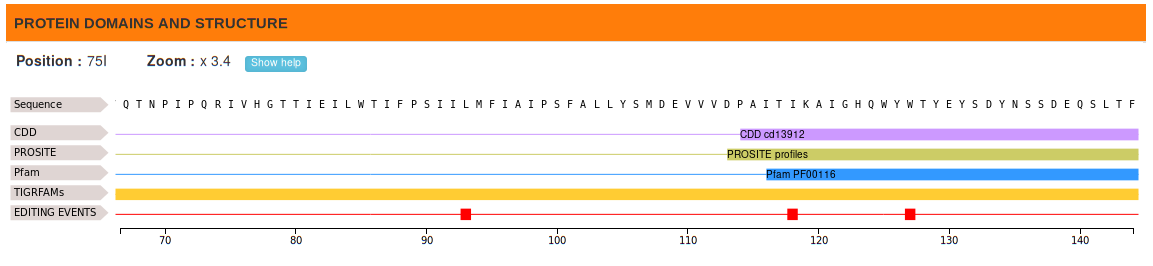
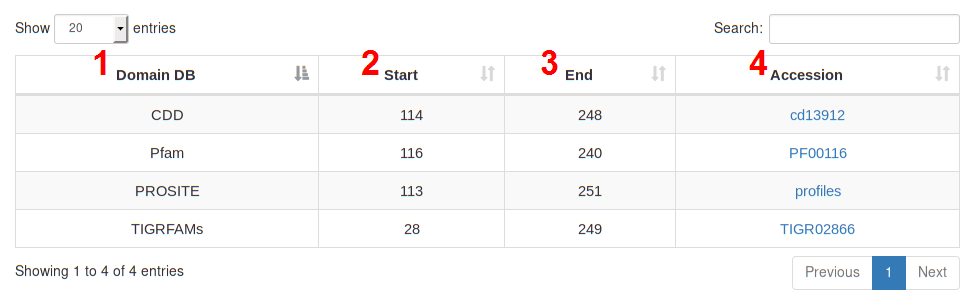
The distribution of RNA editing events along functional domains and predicted protein secondary structures are graphically
rendered by mean of Nextprot® Feature Viewer tool.
Protein domains have been detected using the InterPro engine, while secondary structures have been predicted using the stand-alone version of Spider2 program.
By clicking on the button near a protein sequence,
the PROTEIN DOMAINS AND STRUCTURE PAGE will appear.
This page contains three main sections:
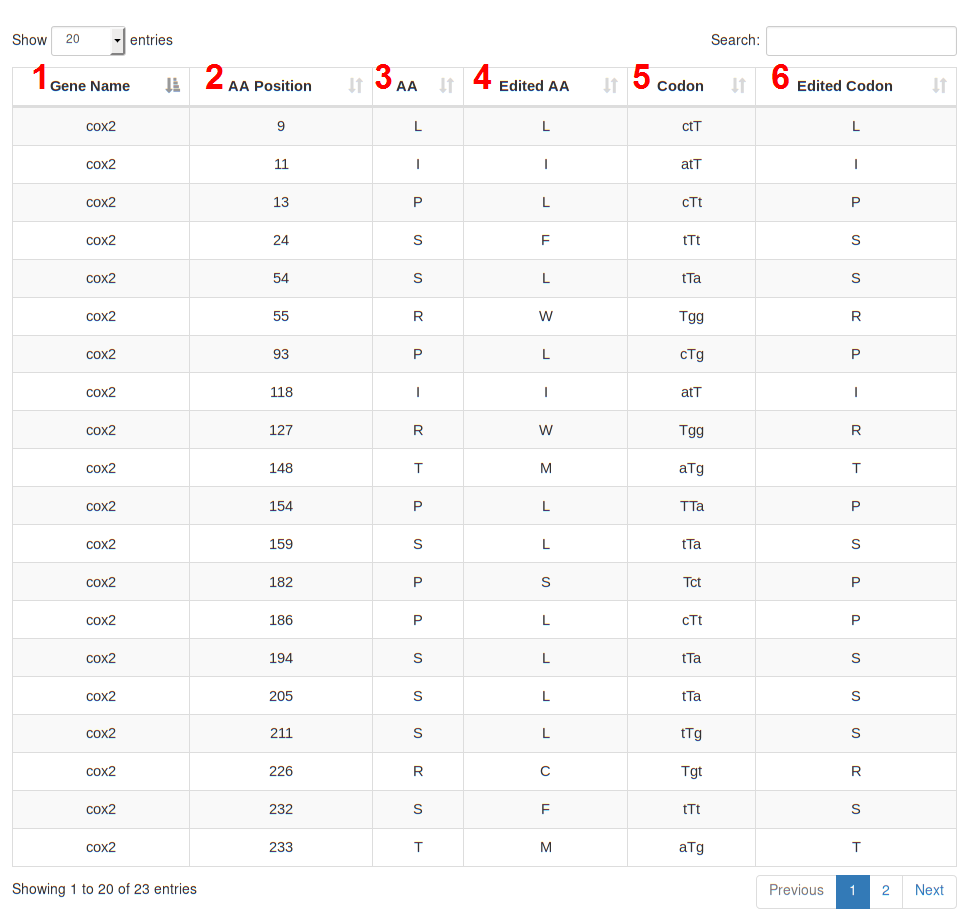
Like the other REDIdb tables, the user can select how many rows wants to show per page. A search filter on the right corner of the tables, can also be used to reduce the rows only to them matching specific criteria.
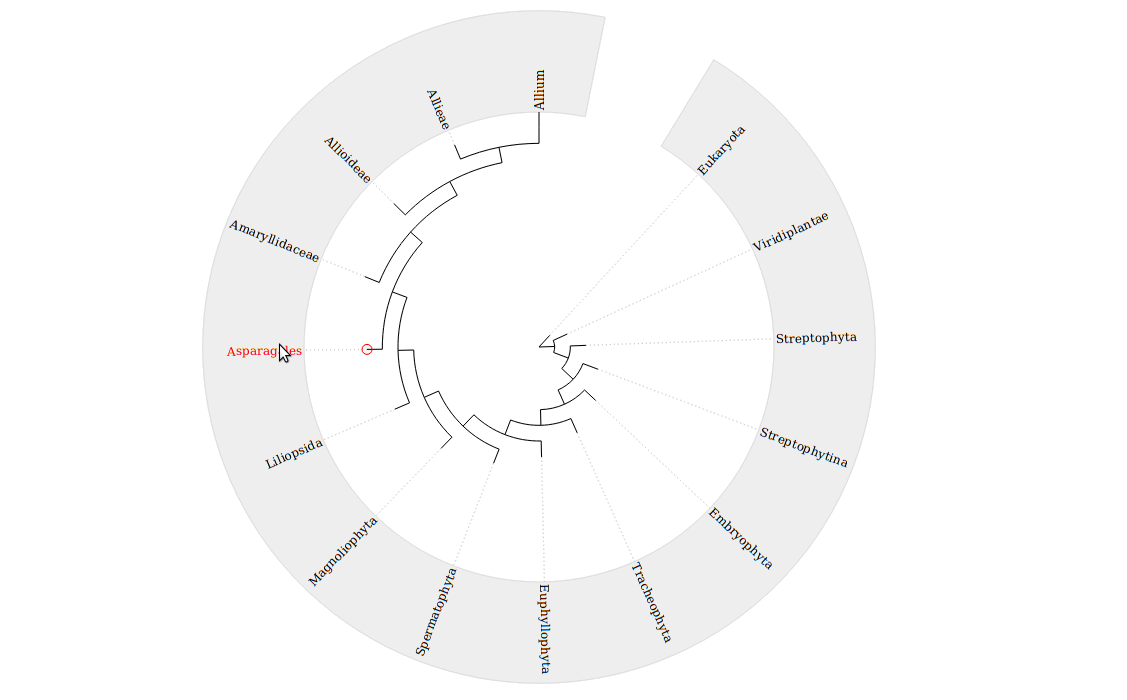
jsPhyloSVG® is a javascript library specifically designed for rendering phylogenetic trees.
Its recent implementation in REDIDB, allows the user to visualize the taxonomy
of each organism in a pleasant and interactive way.
A circular phylogenetic tree based on jsPhyloSVG is displayed by clicking on the
in the Table Report page.
Taxonomy representation follows the Newick tree format. By moving the mouse cursor over a taxonomic rank, the corresponding tree node will appear highlighted in red.
Every complete genome is automatically assembled through a series of custom python scripts
according to the order of genes reported in the genbank flatfile.
If both RefSeq and non RefSeq data are available for the same organism, highest priority is given to RefSeqs.

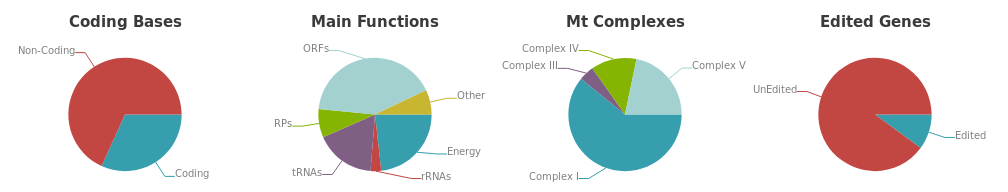
Statistics such as the coding potential of the genome as well as the fraction of edited genes are available in a dedicated section under the genome graph. The section is organized in four main pie charts:
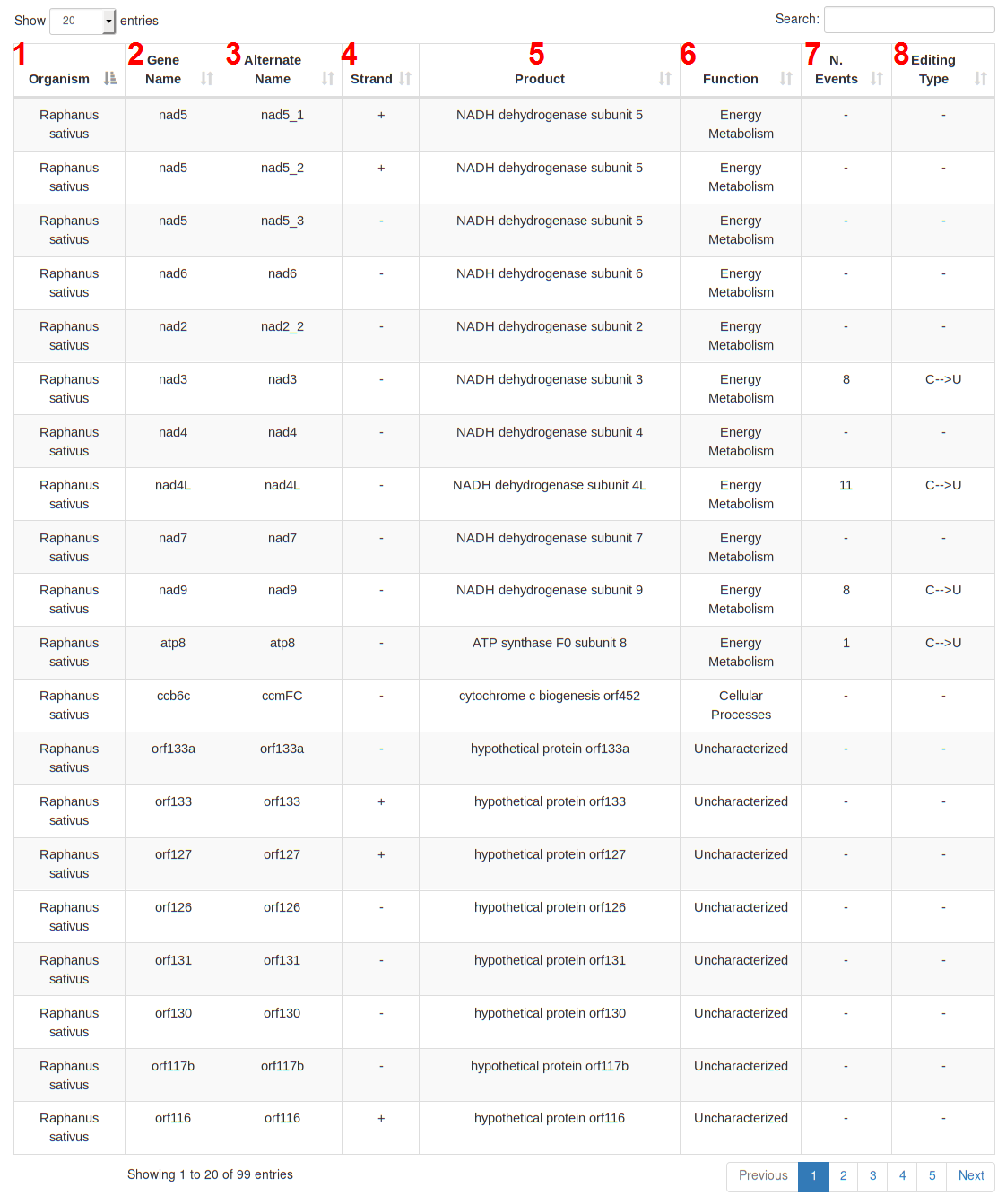
A table detailing each gene is available at the bottom of the genome page.
The table contains the following columns:
Following the pipeline used to generate the previous releases of REDIdb, all the records containing changes due to RNA editing are
derived from GenBank.
The initial search is performed using the query '("RNA edited"[All Fields] OR "RNA editing"[All Fields])’
and positive entries are collected in textual format by using an ad hoc python script.
By using separate scripts, each entry is catalogued as mitochondrial or plastidial, and the corresponding feature table is parsed.
Each genbank record is dissected in its main features (CDS, intron, exon, tRNA, rRNA, misc_feature) using the “genbank” module contained
in the Biopython SeqIO parser (e.g. SeqIO.parse(‘gb.record’,”genbank”)).
This allows the conversion of each record’s feature in a manageable sequence object with its main methods (e.g count, find,
complement, transcribe, translate, extract, ecc.) and qualifiers (gene, product, strand, location, ecc).
A dictionary containing the nucleotide sequence, the genomic strand, the genomic coordinates and the sequence length
is then generated for each type of record feature.
In the specific case of the misc_features fields, under which the editing event are tipically reported,
the associated dictionary contains the annotation (e.g. /note="C to U RNA editing"), the genomic position
and the strand.
Taking advantage of the fact that a misc feature seq objects share the same qualifier with the feature to which
it refers (CDS, tRNA, ecc.), by using this attribute as key of the aforementioned dictionaries,
each editing event is uniquely mapped to its sequence.
Details like pubmed references, taxonomy, are
obtained from each genbank record object's qualifiers generated by the SeqIO parser
(e.g record.annotations['references'], record.annotations['taxonomy'], ecc.).
Due to the lack of control on the Biopython SeqIO nucleotide translation module (expecially in the case of truncated sequences and non standard codon tables)
protein translation and aminoacidic changes are calculated by mean of a custom python module.
Adhoc python code was also written for managing the remaining information appearing in REDIdb
(gene ontologies, protein domains, ecc.).
All the scripts used to generate the REDIdb database are open source and freely accesible upon request (at this page).
From this page you can download: