
ASPICdb Help Guide
ASPICdb is a database tool for alternative splicing analysis that collects the results obtained by ASPIC algorithm (Castrignanò et al. 2006) applied to the complete set of human genes for which a NCBI Reference mRNA sequence and a Unigene cluster are available. In the near future, we plan to include the full gene set of other organisms.
This help page is intended to introduce new users to ASPICdb providing information on the basic retrieval procedures.
If you wish to analyze the splicing pattern of a specific gene just type its Hugo ID in the Search for Gene form shown below:
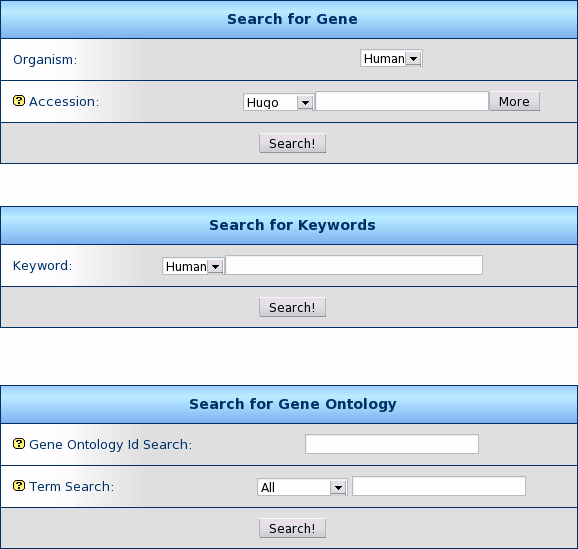
Alternatively, other IDs can be used such as Unigene (e.g. Hs.654481), RefSeq (e.g. NM_000546), Entrez (e.g. 7157), MIM (e.g. 202300).
You can also search for more than one gene, by using
the More option, which opens new text boxes for inputting the names of
additional genes.
The middle panel allows a keyword search (e.g. survinin) whereas the lower panel of the search form is devoted to Gene Ontology searches. You can retrieve genes related to a given GO ID (e.g. GO:0000739) or to GO categories matching text terms related to the biological function, the cellular component or the biological process (e.g. mitochondrion).
The same form appears by clicking on the Search button on the left menu.
If you wish to carry out an Advanced Search just click this item on the left menu, and then, first of all, select genes, transcripts or splice sites depending on the type of search you wish to perform.
Depending on the choice made a new form appears.
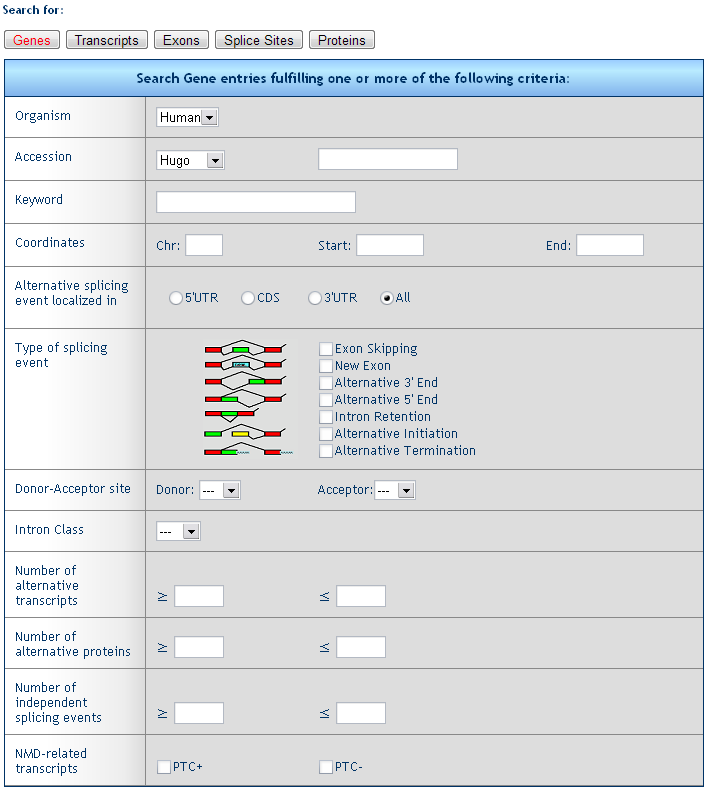
1) Gene retrieval form
The form allows for the search of genes fulfilling one or more criteria
(A)
(B)
Organism: select the species
Accession: retrieve a gene with a given Unigene (e.g. Hs.654481), RefSeq (e.g. NM_000546), Entrez (e.g. 7157), or MIM (e.g. 202300) ID.
Keyword: retrieve all genes characterized by a given keyword (e.g. survivin)
Coordinates: retrieve all genes falling within the given chromosome region.
Alternative splicing events localized in: retrieve genes with alternative splicing affecting the 5UTR, the CDS or the 3UTR
Type of splicing event: retrieve genes with one or more type of splicing event (e.g. exon skipping, alternative 3 end, etc.)
Donor or Acceptor site: retrieve genes containing at least one splice site with the chosen assortment of donor and acceptor splices (e.g. AT/AC).
Intron class: retrieve genes containing U2, U12 or unclassified (ND) introns.
Number of alternative transcripts: retrieve genes generating a specific number (range) of alternative transcripts (e.g. ≥50).
Number of alternative proteins: retrieve genes generating a specific number (range) of alternative proteins (e.g. ≥50).
Number of independent splicing events: retrieve genes with a specific number (range) of independent splicing events (i.e. a specific exon skip can be predicted in more than one alternative transcripts, but it is counted only once).
NMD related transcripts: retrieve all genes encoding (PTC+) or not (PTC-) transcripts containing a premature stop codon.
After the query is completed, a Result Table is shown, like in the example below.
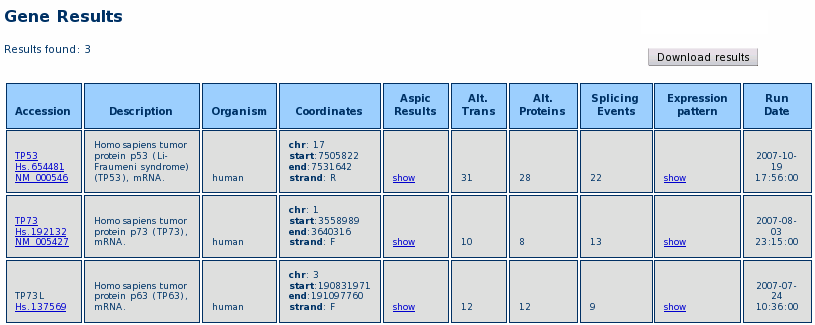
For each gene, the result table reports the Hugo, Unigene and RefSeq IDs, a short description, the chromosome location, a link to the ASPIC results (see ASPIC output section), the number of alternative transcripts, alternative proteins and independent splicing events and the date of the ASPIC run that generated the predictions stored in the database.
2) Transcript retrieval form
The form allows for the search of single transcripts fulfilling one or more criteria.
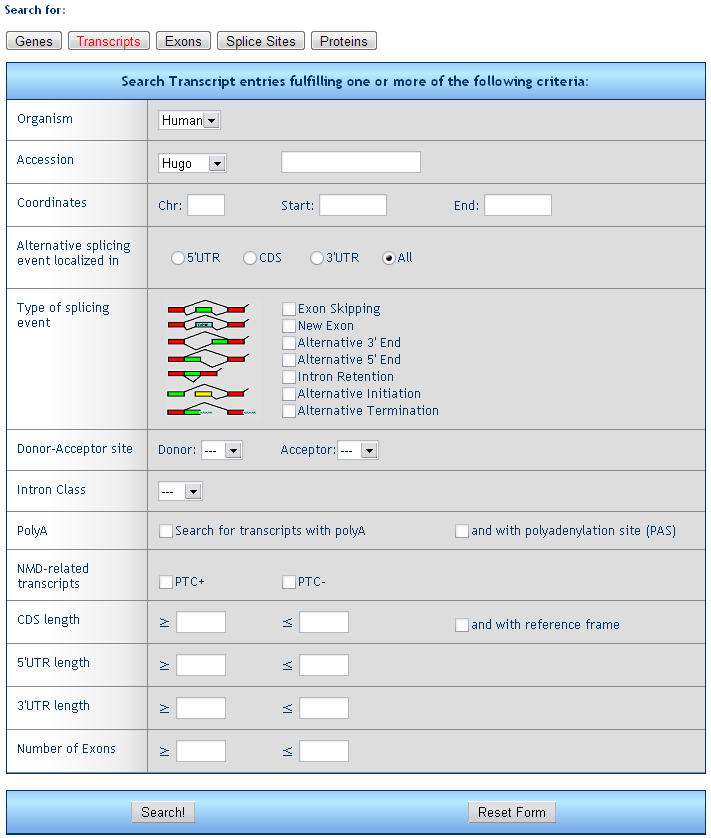
Organism: select the species (only human is available to date)
Accession: retrieve all transcripts encoded by a gene with a given Unigene (e.g. Hs.654481), RefSeq (e.g. NM_000546), Entrez (e.g. 7157), or MIM (e.g. 202300) ID.
Signature ID: Unique signature ID assigned according to Riva and Pesole (2009) (e.g. 4305f45b95:11)
Coordinates: retrieve all transcripts from genes falling within the given chromosome region.
Alternative splicing events localized in: retrieve transcipts with alternative splicing affecting the 5UTR, the CDS or the 3UTR
Type of splicing event: retrieve transcripts with one or more type of splicing event (e.g. exon skipping, alternative 3 end, etc., all events are referred to the reference transcript)
CAGE tags: retrieve transcripts whose initiation site is supported by CAGE (Cap Analysis of Gene Expression) tags produced by RIKEN in Japan
Donor or Acceptor site: retrieve transcripts whose precursor mRNA contains at least one splice site with the chosen assortment of donor and acceptor splices (e.g. AT/AC).
Intron class: retrieve transcripts derived from a gene containing U2, U12 or unclassified (ND) introns.
PolyA: retrieve transcripts containing a polyA tail and a poly-adenylation signal (e.g. AAUAAA).
NMD related transcripts: retrieve all transcripts containing (PTC+) or not (PTC-) a premature stop codon.
CDS length: retrieve all transcripts with an annotated CDS in the required length range.
5'UTR/3'UTR length: retrieve all transcripts with an annotated 5' or 3'UTR in the required length range.
Number of exons: retrieve all transcripts generated by the concatenation of a number of exons in the required length range.
After the query is performed a Result Table is shown, like in the example below.
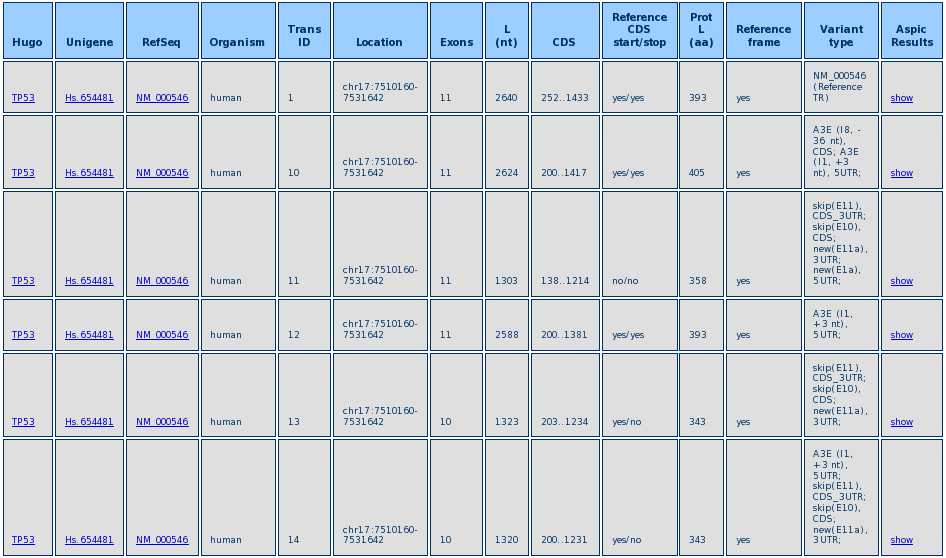
For each transcript, the result table reports the Hugo ID of the relevant gene, the Unigene and RefSeq IDs, the chromosome location, the number of exons, the transcript length, the position of the CDS, the protein length, the usage or not of the reference transcript start/stop codons and reading frame, and a link to the ASPIC results (see ASPIC output section).
3) Exon retrieval form
The form allows for the search of single exons fulfilling one or more criteria.
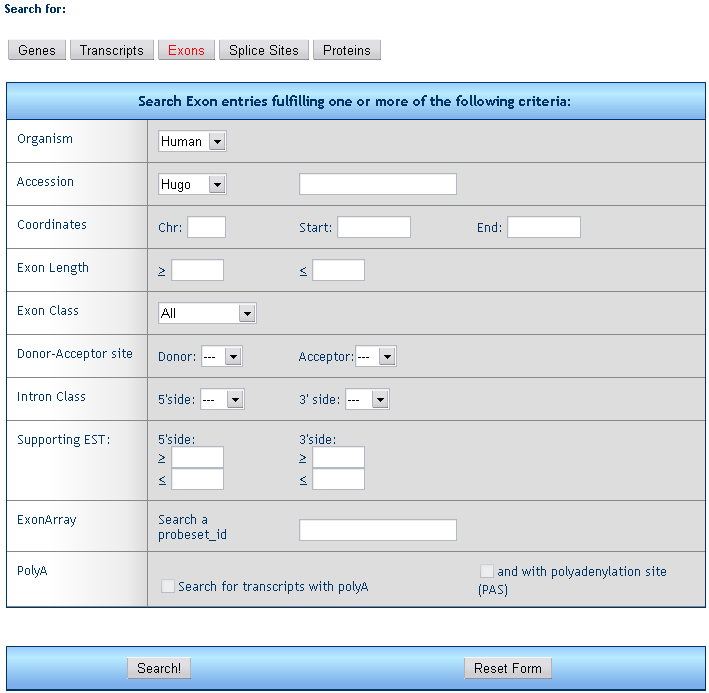
Organism: select the species
Accession: retrieve all exon from genes with a given HUGO (e.g. TP53), Unigene (e.g. Hs.654481), RefSeq (e.g. NM_000546), Entrez (e.g. 7157), or MIM (e.g. 202300) ID..
Coordinates: retrieve all exons falling within the given chromosome region.
Exon length: retrieve all exons within the given length range.
Exon class: retrieve all exons belonging to the given class (e.g. initial, internal, terminal).
Donor-Acceptor site: retrieve all exons flanked by specific donor and acceptor sites.
Intron class: retrieve all exons with specific intron types on 5. and 3. side.
Supporting ESTs: retrieve all exons suppoted by a given range of ESTs on both 5. and 3. side.
ExonArray: retrieve all exons mapping a given Affimetrix Exon Array probeset (e.g. 3743928)
PolyA: retrieve all exons containing a polyA tail and a poly-adenylation signal (e.g. AAUAAA)
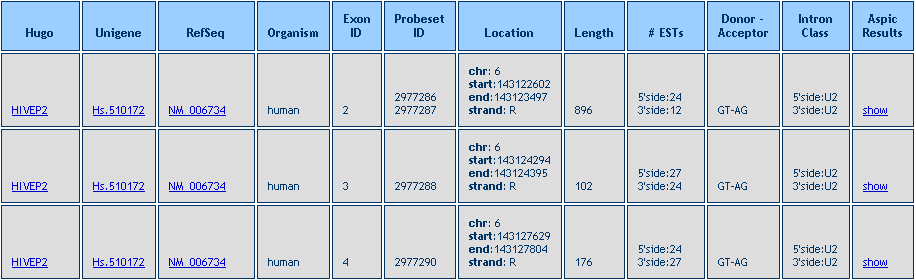
After the query is performed a Result Table is shown, like in the example below.
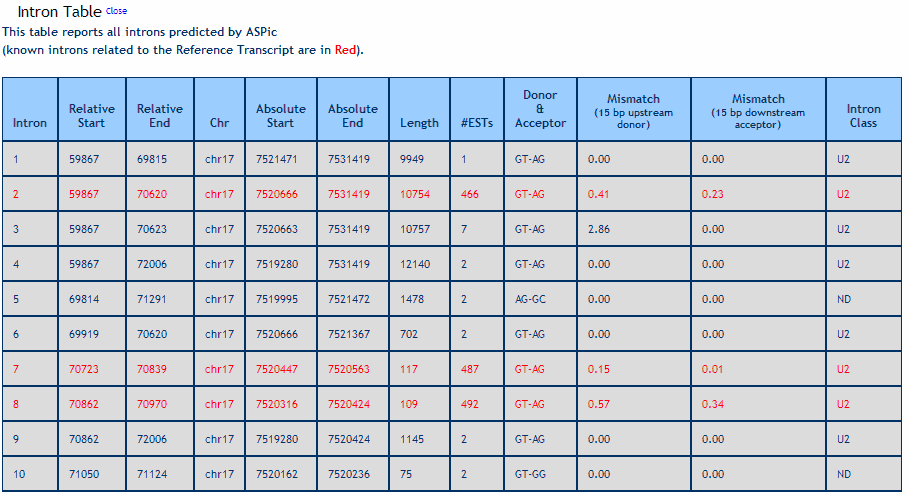
4) Splice site retrieval form
The form allows for the search of introns fulfilling one or more criteria
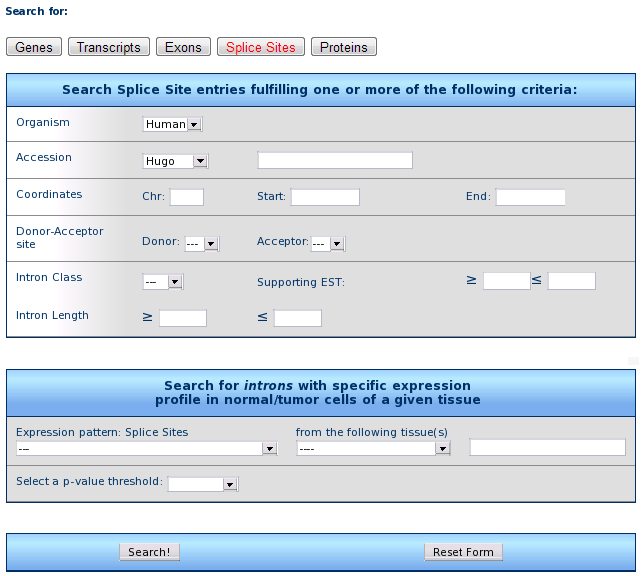
Organism: select the species (only human is available to date)
Accession: retrieve all introns included into a gene with a given Unigene (e.g. Hs.654481), RefSeq (e.g. NM_000546), Entrez (e.g. 7157), or MIM (e.g. 202300) ID.
Coordinates: retrieve all introns from genes falling within the given chromosome region.
Donor or Acceptor site: retrieve introns with the chosen assortment of donor and acceptor splices (e.g. AT/AC).
Intron class: retrieve U2, U12 or unclassified (ND) introns, whose prediction also supported by a given number (range) of ESTs (you can specify either parameter, or both).
Intron length: retrieve all introns in the required length range.
After the query is performed a Result Table is shown, like in the example below.
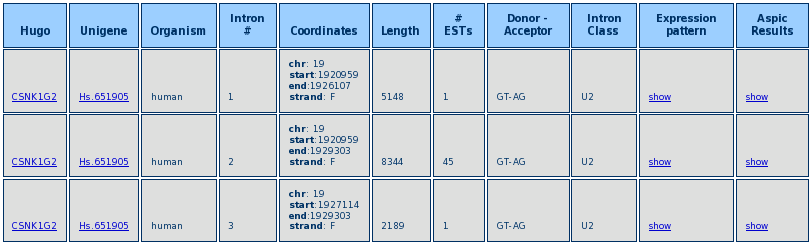
The Table shows the Hugo and Unigene IDs, the intron number (following the progressive numeration in the ASPIC output) the intron length, the number of supporting ESTs, the donor and acceptor dinucleotides, the intron class and a link to the ASPIC output (see the section on the ASPIC output).
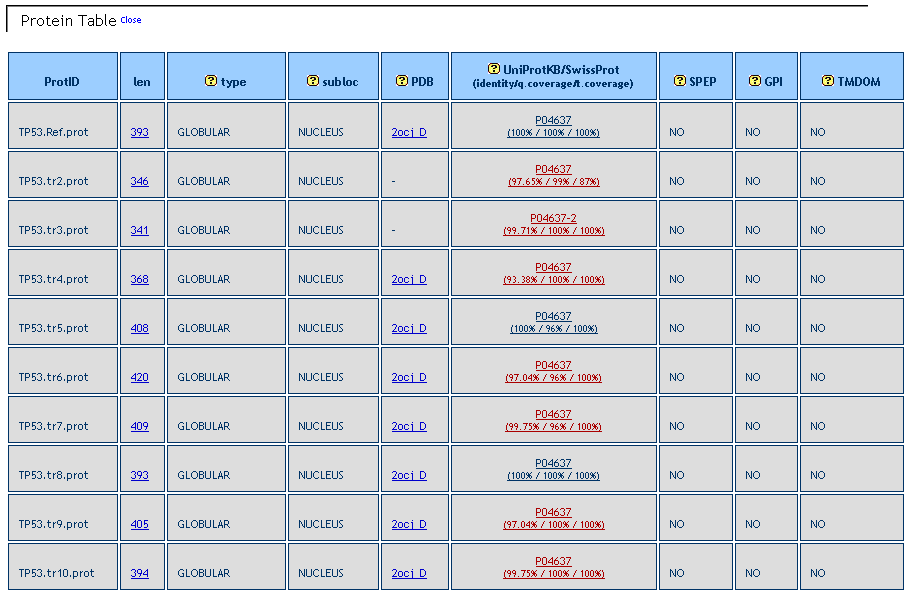
5) Protein retrieval form
The form allows for the search of proteins fulfilling one or more criteria.
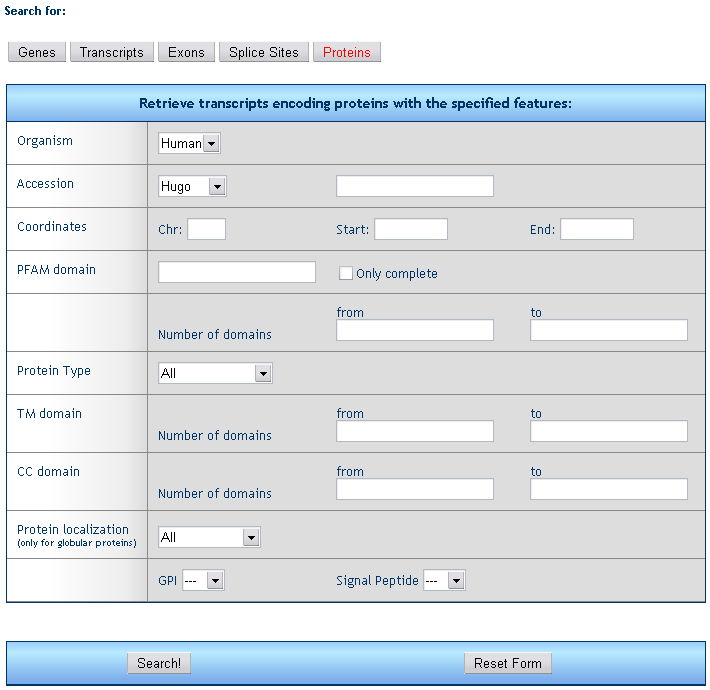
Organism: select the species (only human is available to date)
Accession: retrieve all proteins encoded by genes with a given HUGO (e.g. TP53), Unigene (e.g. Hs.654481), RefSeq (e.g. NM_000546), Entrez (e.g. 7157), or MIM (e.g. 202300) ID..
Coordinates: retrieve all exons falling within the given chromosome region.
PFAM domain: retrieve all proteins containing a specific type and number of PFAM domains, evenually required to be complete.
Protein type: retrieve globular or transmembrane proteins.
TM domain: retrieve proteins containing a given range number of transmembrane domains.
CC domain: retrieve proteins containing a given range number of coiled coil domains.
Protein localization: retrieve globular proteins with a specific subcellular localization and/or containing a GPI anchor propeptide or a Signal Peptide.
After the query is performed a Result Table is shown, like in the example below.
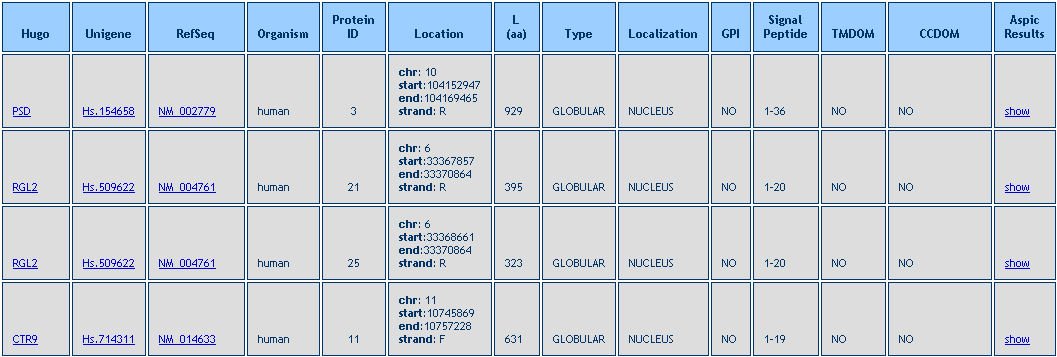
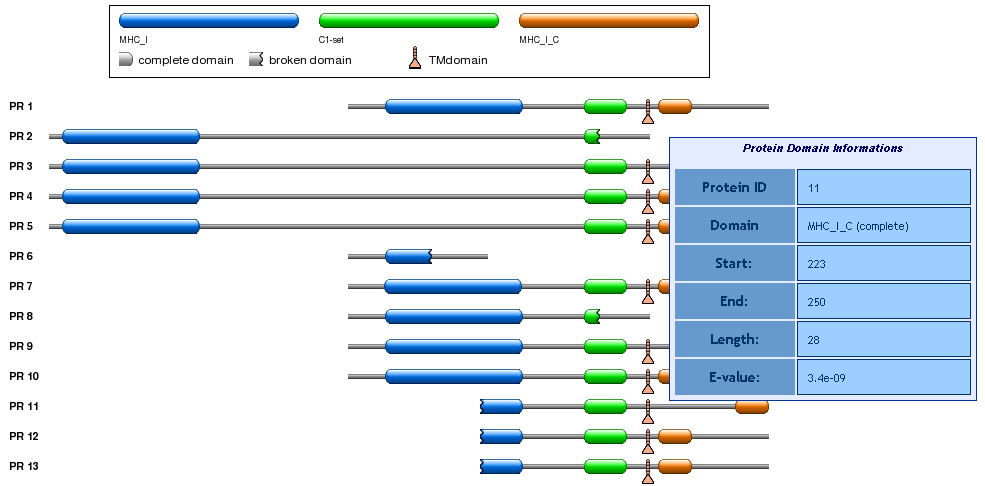
By clicking the show link (last column in the Result Table) a set of tabular and graphical views are shown.
For each alternative protein all annotated functional domains are reported, as shown in the image below.
For each domain a tooltip provides detailed information (i.e. domain name, coordinates, length, e-value).
Data and sequence download
After a query has been accomplished at the gene (A), transcript (B), exon (C), splice sites (D) or protein (E) level you can download specific sets of sequences in multiFASTA format (e.g. genes, transcripts, proteins, 5UTRs, coding sequences, 3UTRs, introns) as well as sequence regions surrounding splice site boundaries. Such sequences may subsequently used for customized sequence analyses such as pattern matching and discovery approaches aimed at the identification of splicing regulatory elements.
(A) Gene Download Form
(B) Transcript Download Form
(C) Exon Download Form
(D) Splice Sites Download Form

(E) Protein Download Form
The sequence header univocally labels each extracted sequences including the gene name, the transcript or intron number (as in the Aspic output), etc.
>TP53 | Intron2 | donor site
TTCCTCTTGCAGCAGCCAGACT
>TP53 | Intron2 | acceptor site
CCTGGATTGGGTAAGCTCCTGACTGAACTTGA
>TP53 | Intron6 | donor site
TCCCAGCATTTCGGGAGGCTGA
>TP53 | Intron6 | acceptor site
CCCAGCACTTTGGGAGTCGGAGGCGGGAGGAT
As the sequence file generation may be time consuming you have to provide a valid e-mail address to which he will receive notification that the requested file in zipped format is available for downloading, together with a link for its download.
Aspic Output
The ASPIC output is organized in 8 sections: 1) Gene Information; 2) Gene Structure View; 3) Predicted Transcripts; 4) 4) Transcript Table; 5) Predicted proteins; 6) Protein Table; 7) Predicted Splice Sites; 8) Intron Table.
Gene Information. Provides a summary of the submitted input and information retrieved by the ASPic engine (e.g. chromosome number, start, end, strand, etc.). Links in this section allow the user to view or download input sequences.
Gene Structure View. Provides a schematic graphical view of the gene structure (RefSeq exons in blue, novel exons in red) and of the predicted introns (numbered progressively). Known and novel introns are shown as blue and red lines, respectively. Fuzzy introns (i.e. introns with limited support by EST sequences and flanked by non-canonical splice sites) are shown as dashed lines. The structure of the RefSeq gene is shown in red.
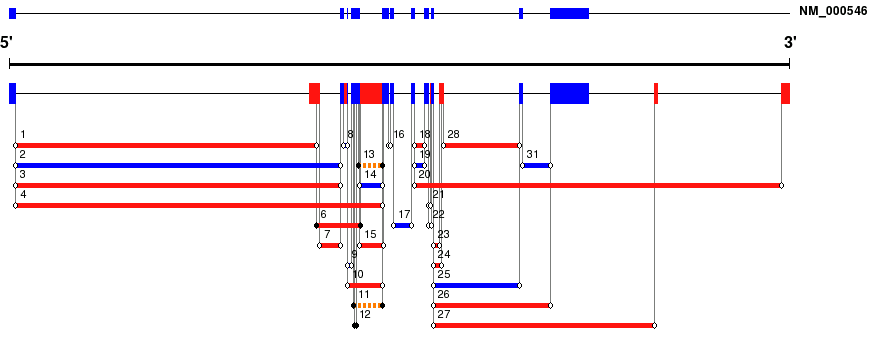
Predicted Transcripts. This view provides a graphical representation of the assembled transcripts. Only the most reliable introns (i.e. those supported by a perfectly aligning EST with canonical donor and acceptor splice sites, or those supported by two or more ESTs) are used for the assembly process. The Predicted Transcripts View shows the overall exon-intron scheme and also reports the annotation for 5'UTR, CDS, 3'UTR, premature stop codons (PTC) and polyA site. The first transcript used as reference, corresponds a NCBI RefSeq transcript, and it is named from the Hugo ID with the ".Ref" extension (e.g. TP53.Ref). If more RefSeq transcripts are annotated for the same gene, is chosen the longer one with the highest number of exons. A 3' terminal arrow marks polyadenylated transcripts that may also contain a polyA site (AAUAAA or its accepted variants) and a hyphen marks the position of mapping CAGE tags which identify transcription initiation sites. Alternative transcripts containing ORFs longer than 100 codons are annotated as coding mRNAs, whereas transcripts containing ORFs shorter than 100 codons are labeled "Unannotated RNA".
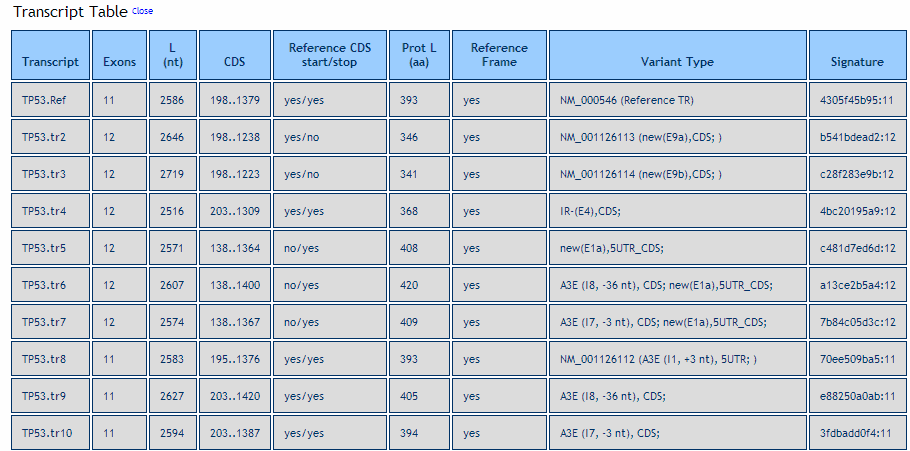
Transcript Table. Shows the general features of all alternative full-length transcripts. It includes information about: the length, the number of exons, the putative location of the CDS, the occurrence of start/stop codons in the reference transcript, the frame sharing with reference mRNA, the length of the putative encoded protein and the transcript variant type. The variant type column reports - for each full-length transcript - the type of splicing event (e.g. alternative 5 or 3 end, exon skipping, etc.), the affected exon or intron as well as its location in the coding and/or untranslated regions of the transcript. Splicing variants are labeled with respect to a reference transcript (the longest transcript in the RefSeq database, see http://www.ncbi.nlm.nih.gov/RefSeq/) if available, or by the longest transcript. The CDS annotation is done accoording to the following procedure. If the transcript isoform totally overlaps or includes a known mRNA (e.g. a RefSeq entry with a CDS linked to a Swissprot/Uniprot curated peptide) the RefSeq annotation is taken in ASPICdb. For the other assembled .novel. transcripts (i.e. those listed in the .predicted transcripts. section of the output, not coinciding with the known RefSeq transcripts) a fully automated reliable prediction of ORFs able to cover every case is almost impossible to obtain without any human correction or experimental validation. Therefore, in order to assess correctly the most common situations we introduced the following criteria: i) if the transcript isoform contains the start codon of the RefSeq transcript then the CDS is annotated as the ORF starting at this ATG, unless a longer ORF in-frame with the RefSeq annotated protein is found; ii) if the transcript does not contain the start codon of the RefSeq transcript (and thus may be potentially truncated) the longest ORF is annotated as the CDS if longer that 300 nt. In this case, no ORF is annotated in shorter than 300 nt.
Used abbreviations for splicing events are:
A3E, A5E: alternative 5 or 3ends (the affected intron number and the length difference with respect to the reference transcript is shown in brackets, e.g. I8, -36nt);
skip: exon skip (the reference exon number is shown in brackets, e.g. E8);
New: novel exon(s) not present in the reference transcript (e.g. E(9a), means a new exong after exon 9 of the reference transcript);
Init: alternative starting exon(s) (e.g. E(0a), means an alternative initiation exong before exon 1 of the reference transcript);
Term: alternative termination exon (in brackets the exon number, e.g. E8);
IR: intron retention (IR- or IR+ are used depending on the intron is present or not in the reference transcript; the length difference with respect to the reference transcripts related to the IR event is also reported in the brackets);
After each splicing event is also reported the affected location in the reference mRNA: 5UTR (5UTR); CDS; 3UTR (3UTR); uCDS (5UTR and CDS); CDSu (CDS and 3UTR);
GTF. Provides a full textual output of ASPic including: i) absolute intron coordinates and IDs of supporting ESTs; ii) intron composition of assembled transcript isoforms; iii) absolute exon coordinates; iv) exon composition of assembled transcript isoforms; v) nucleotide sequence of assembled transcript isoforms in FASTA format (header line reports IDs of polyadenylated transcript).
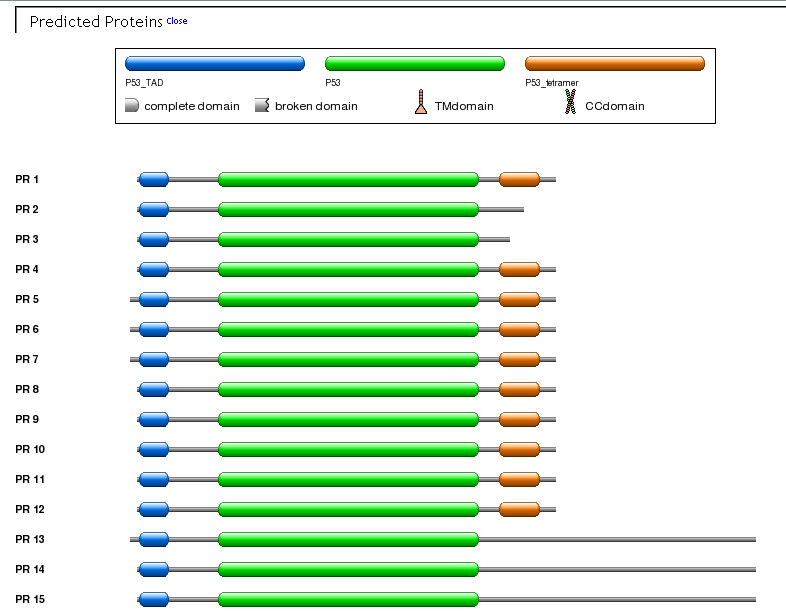
Predicted proteins. This view provides a grafical representation of encoded proteins showing the position of PFAM, transmembrane (TN) and coiled-coil (CC) domains. If you put the mouse pointer an a domain specific information will be visualized into a popup window.
Using FuzzPro all human Mass Spectrometry (MS) derived peptides from PeptideAtlas had been aligned against protein isoforms collected in ASPicDB. Of the total 72,396 peptides (build 7/2010), we mapped 66,765 peptides on 138,731 different protein isoforms from 7,612 genes. These results can provide an experimental validation of ASPIC prediction data. Matching regions are graphically shown on both Transcript and Protein View (see the Figure), with more detail reported in the relevant tooltips.
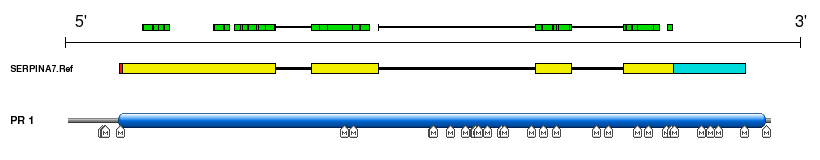
Protein table. Lists the features of the alternative proteins. It includes information about length, protein type (globular or transmembrane), subcellular localization, matching % identiy and % coverage ith UniprotKB/SwissProt entries, occurrenco of signal peptides, GPI anchor propeptides and transmembrane domains.
Predicted Splice Sites. This view shows the alignment between the genomic sequence and the transcribed sequences near the predicted intron boundaries (15 nt upstream and downstream of intron boundaries). The donor and acceptor quality score as well as the classification as U2 or U12 introns is done according Sheth et al. (2006). A branch site is searched within 200 bb from the acceptor site and annotated when its score is ≥0.75.

Intron Table. Reports the relative and absolute coordinates of each detected intron, the number of supporting ESTs, the intron length, donor and acceptor splice sites, the alignment quality near intron boundaries (expressed in Mismatch %) and the intron class (U2 or U12). Known introns related to RefSeq transcripts are in red.
Bibliography
Castrignano T, Rizzi R, Talamo IG, De Meo PD, Anselmo A, Bonizzoni P, Pesole G. ASPIC: a web resource for alternative splicing prediction and transcript isoforms characterization. Nucleic Acids Res. 2006 Jul 1;34 (Web Server issue):W440-3.
Wang Z, Lo HS, Yang H, Gere S, Hu Y, Buetow KH, Lee MP. Computational analysis and experimental validation of tumor-associated alternative RNA splicing in human cancer. Cancer Res. 2003 Feb 1;63(3):655-7